Angelman syndrome is a rare genetic disorder. It consists of a defect in the structure of chromosome 15, which is formed during the process of cell division during embryogenesis. Such an anomaly leads to the birth of children with characteristic abnormalities. Children are lagging behind in mental and physical development, but at the same time they are characterized by optimism and cheerfulness, which is why some literary sources use the term “angel syndrome” to refer to the disease. In addition to these deviations, patients with the pathology suffer from hypotonicity, increased activity and speech disorders. There is no specific treatment for Engelman syndrome. Moreover, people with this pathology live on average as long as healthy people. Therapy is symptomatic in nature and is aimed primarily at adequate socialization.
History of the study of the disease
The disease is named after Harry Angelman, a British pediatrician who first differentiated this syndrome in 1965. Then it was called happy puppet syndrome, but today this term is not used, as it was considered disparaging.
Dr. Angelman had treated several children with similar symptoms and suspected a common diagnosis. It was not possible to prove the diagnosis and obtain accurate data at that time due to the lack of technologies that are available today. The doctor reflected his guesses in an article entitled “Children of Puppets.”
At that time, the publication did not arouse much public interest, and was soon forgotten. It was remembered again in the eighties, when the necessary research methods appeared. Scientists have found that most children suffering from the syndrome are missing a small part of chromosome 15.
Treatment in Italy
Russian Medical Server / Treatment in Italy / Center for the treatment of rare diseases in Milan / Angelman syndrome - treatment in Italy
Angelman syndrome is a fairly rare disease caused by chromosomal abnormalities and occurs in approximately 1 in 10,000 newborns. Children with this defect experience delayed mental and neurological development from the first years of life.
Since we are talking about a genetic defect, there is no drug treatment for it, however, there are procedures that can improve the quality of life of patients diagnosed with Angelman syndrome. For example, from early childhood, experts recommend giving the patient massage and physical procedures to strengthen the body.
With proper care, people with this disorder can live into adulthood and care for themselves, although they may never be completely independent.
The disease is named after Harry Angelman, a pediatrician from Great Britain who first diagnosed the above-mentioned disorder in 1965, calling patients “doll children.”
As a doctor at Warrington Hospital, the medic cared for three young disabled patients with different symptoms that initially seemed to be the result of different diseases. However, the specialist felt a commonality between them. Based on clinical studies, Angelman made a general diagnosis, but could not prove it at a more precise level, since the laboratory equipment of that time did not allow such complex genetic studies. For this reason, Angelman was in no hurry to publish his thoughts in medical journals.
The pediatrician was given confidence in these guesses by the painting “Boy Puppet”, painted in oil, which he saw in the Italian museum in the city of Verona. The picture shows a laughing boy. He reminded the doctor of all three of his patients so much that Angelman decided to write one general article about them, called “Children Puppets.”
The scientific work was published in 1965. She aroused a little interest, but was soon quickly forgotten about. Doctors return to the article already in the eighties, when references to the disease began to appear in the medical community. Only by 1987, scientists were able to establish a pattern that most children with the syndrome are missing part of chromosome 15.
Due to the fact that the name of the diagnosis “happy puppet syndrome” seemed scary and humiliating to many parents, it was later renamed using the name of the scientist Angelman.
The syndrome is diagnosed by genetic analysis (chromosome 15), recommended for newborns with reduced muscle tone (hypotonicity), delays in the development of gross motor skills and speech development. Parents and doctors should pay attention to cases of small tremors, chaotic, jerky movements of the limbs, gait with stiff legs; in some cases, a specific facial expression, too frequent laughter.
Possible analysis methods: fluorescence in situ hybridization process, DNA methylation in the 15q11-q13 region, imprinting center mutation analysis, direct mutation analysis of the UBE3A gene.
Genetic analysis allows us to identify a number of pathologies that may be detected in an unborn child.
Chromosomal diseases are quite severe, and treatments for them have not yet been found. For this reason, parents must prepare for what the future holds. In some cases, genetic testing for chromosomal diseases is necessary in order to decide whether to terminate a pregnancy.
Angelman syndrome is a congenital genetic abnormality; Currently, specific methods for its treatment have not been developed, but some therapeutic measures improve the quality of life of people with the syndrome. Infants with hypotonia should receive massage and other special therapies (physical therapy).
It is recommended to use special methods of child development, classes with a speech therapist and speech pathologist. Sleep disturbances are corrected by prescribing mild sleeping pills. Dr. Wagstaff (USA) believes that administering 0.3 mg melatonin 30 minutes to 1 hour before bed improves sleep in patients with Angelman syndrome. Stool disorders are regulated by the administration of mild laxatives.
Seizures are treated in the same way as epilepsy. Children with Angelman syndrome often experience more than one type of seizure. Electroencephalography is indicated.
Undesirable behavior. Dr. Charles Williams (Gainesville, Florida), who works primarily with autistic children, notes common behavior patterns among autistic children and children with Angelman syndrome: marked self-stimulation, impulsivity, compulsive, repetitive movements, interest in inappropriate objects, and difficulty in communicating with other people.
Doctors show that for autistic children, intravenous injections of the hormone secretin (found in the pancreas) successfully reduce unwanted behavior and promote good levels of sociability and communication skills; Perhaps medicine will come to the use of secretin to correct the behavior of children with Angelman syndrome.
! Despite the fact that many of the diseases described in this section are considered incurable, the Center for the Treatment of Rare Diseases in Milan is constantly looking for new methods. Thanks to gene therapy, it has been possible to achieve outstanding results and completely cure some rare syndromes.
Contact a consultant on the website or leave a request - this way you can find out what methods Italian doctors offer. Perhaps this disease has already been treated in Milan.
+7 (925) 50 254 50 – urgent treatment in Italy
REQUEST TO THE CLINIC
Pathogenesis and causes
This defect occurs due to deletion (loss of a piece of genetic material) of a segment of chromosome 15. Other causes are often cited as unipaternal disomy (inheritance of two paternal copies of a chromosome, instead of copies from both parents), translocation or mutation of a single gene.
The syndrome can also appear as a result of a mutation in a gene involved in ubiquitin metabolism.
Most patients with this pathology do not have a family history of genetic abnormalities. However, for a certain percentage of patients this syndrome is inherited. The risks become higher if the parents have chromosomal abnormalities. An interesting fact is that if a child with this syndrome is born in a family, then the probability of having another child with the same pathology is 1%.
The appearance of Angelman syndrome is quite spontaneous, and almost any family can have a child with this disease. There are no specific statistics on the number of children with this syndrome.
According to various estimates, the number of newborns with doll syndrome ranges from one in ten thousand to one in twenty thousand. Scientists suggest that in reality this number is much higher.
Diagnosis of Angelman syndrome
Diagnosis of the condition associated with happy doll syndrome is made based on a detailed review of the medical history, a thorough clinical assessment, and identification of the characteristic manifestations of the disorder.
Approximately 80% of cases can be confirmed using a specific blood test based on DNA methylation.
This method allows you to detect a failure of the genetic program, but does not make it possible to determine whether it occurred, for example, due to a chromosome deletion or a defect in the imprinting center.
Genetic mutations increase the risk of developing the disease
The appearance of Angelman syndrome is associated with the presence of various chromosomal abnormalities in the parents of the unborn child. Among such deviations are usually called:
- chromosome trisomy - the presence of one or more extra chromosomes in the chromosome set;
- inversion - a turn of one of the chromosome sections by 180 degrees, while part of the chromosome is skipped, and the genes are arranged in the opposite order;
- microdeletion , which is the result of rearrangement of the Y chromosome and the exchange of sections between chromosomes, a small number of chromosomes is observed, and one of the genes may be missing;
- deletion – lack of one of the chromosome sections;
- translocation - transfer or attachment of a section of one chromosome to another chromosome;
- duplication - copying part of chromosomes, resulting in excess genetic material;
- ring chromosome - there is no genetic material at the ends of the chromosome, while the newly formed ends are connected in the form of a ring.
Gene mutations that can cause the syndrome
Causes
It is known that pathology has a genetic basis. The risk of developing a defect increases if one of the parents suffers from chromosomal abnormalities. In this case, the father or mother can only be carriers of diseases. The exact causes of disruption of cell division processes in the embryonic period are unknown. Presumably, drug addiction, alcohol abuse or potent drugs during pregnancy can have a negative impact on the growth and development of the fetus.
Symptom groups
Angelman syndrome is usually diagnosed between the ages of 3 and 7 years, when symptoms become severe. A newborn child with the syndrome does not differ from healthy children, however, during the first months of life, problems with feeding appear, children slowly gain weight, problems with sleep, and delays in the development of motor skills are observed.
All symptoms of parsley syndrome are divided into several types. They have a relationship with the neuralgic, mental and physical condition of the patient:
- Physical symptoms include vision problems (strabismus, optic atrophy), tongue protrusion, wide mouth, large distance between teeth, head size proportionately smaller relative to the size of other parts of the body, albinism and hypopigmentation, inflexion of the leg joints when walking (hence the comparison with puppets), scoliosis.
- Neurological symptoms include tremors in the limbs, a range of sleep disorders, hysterical fits, the intensity of which is high at the age of three and gradually decreases as they grow older, problems in the communication process (delayed speech development), hyperactivity and attention deficit.
- Psychological symptoms are usually expressed in overly emotional behavior (always happy appearance, friendliness), severe mental retardation.
Treatment
Angelman syndrome is an incurable genetic abnormality. Currently, medical scientists are actively developing effective therapeutic techniques. If the syndrome was diagnosed during fetal development, experts recommend terminating the pregnancy. A newborn sick child requires careful care, care and highly qualified therapy.
Symptomatic treatment helps alleviate the condition of patients with marionette syndrome. They are prescribed medications and non-drug procedures:
- Anticonvulsants reduce the frequency and severity of seizures. Patients are prescribed several anticonvulsants at the same time, since attacks are characterized by several types of seizures. The most popular drugs include: Valproic acid, Convulex, Lamotrigine, Clonazepam. These drugs prevent seizures, improve the mood and mental state of patients.
- Vitamin therapy is the intake of vitamins B, C, D and E. Treatment with vitamins should only be carried out as prescribed by a doctor, since they reduce the effectiveness of antiepileptic drugs.
- Sleeping pills that improve sleep in easily excitable patients - Melatonin, Diphenhydramine.
- When problems with digestion and stool occur, patients are prescribed laxatives – “Senade”, “Slabilen”, “Fitolax”, pre- and probiotics – “Hilak Forte”, “Linex”, “Bifiform”.
- Hormonal therapy is indicated in extreme cases when it is impossible to correct the behavior of patients in other ways. They are injected with the hormone secretin, which normalizes digestion and has a positive effect on patients’ attention, as well as oxytocin, which improves the child’s cognitive abilities, memory, and behavior. A similar hormone treatment regimen was developed by American scientists for the treatment of children with autism.
- For Angelman syndrome, behavioral therapy, work with a psychologist, speech pathologist and speech therapist are indicated.
- Physiotherapeutic procedures help cope with muscle hypotonia and joint problems. Doctors usually prescribe paraffin baths, electrophoresis, and magnetic therapy. Exercise therapy, professional massage, and aqua gymnastics in cool water help patients stand more confidently on their feet.
- To get rid of hypersalivation, drugs are used that inhibit salivation and surgical interventions aimed at reimplantation of the salivary ducts.
- Traditional medicine, like homeopathic medicines, are weakly effective in treating Angelman syndrome, but are absolutely safe. A collection based on peony, licorice and duckweed reduces the frequency of convulsive attacks, a decoction of lavender and an aqueous infusion of motherwort have a general calming effect.
Clinical picture in detail
Different patients may exhibit different symptoms. This difference depends on the degree and type of chromosomal abnormality.
Signs can be divided according to the frequency of their manifestation in children with Angelman syndrome:
- Symptoms that appear in all patients . These include severe functional developmental delay, behavioral deviations (laughter and smiling for no reason, a state of happiness, increased excitability, decreased concentration). Also, absolutely all patients experience impaired motor functions, impaired balance, tremors, a predominance of nonverbal skills over verbal ones, and speech disorders.
- Symptoms characteristic of 80% of patients . Stunted growth leading to disproportion of the head relative to other parts of the body. This often leads to the development of microcephaly. Children under three years of age often experience epileptic seizures, then they become less frequent or disappear altogether. Electroencephalogram results are often abnormal, with low-level waves having increased amplitude and temporal dynamics.
- Symptoms that occur in less than 80% of patients . Such manifestations include strabismus, decreased ability to control tongue movements, problems with swallowing, hypopigmentation of the eyes and skin, albinism. Many patients experience increased activity of tendon reflexes. In early childhood, many people have problems with eating and sleeping. Manifestations also include drooling, tongue protruding, and constant thirst. External signs also include the presence of a flat back of the head and smooth palms.
As you grow older, the manifestations of the syndrome change. Sleep disorders, hyperactivity, and seizures are less common. Adults with Angelman syndrome appear younger than their peers.
Puberty occurs a little later than in individuals who do not have this pathology. Patients can have their own children, but there is a high risk of transmitting the disease to their offspring.
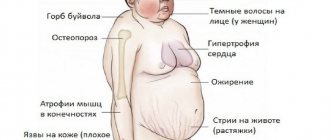
Many adult patients suffer from uncontrollable urination. Often there are difficulties with motor skills, which leads to the need to wear clothes without zippers and buttons. Among adult patients, there is a problem of excess weight, so it is necessary to monitor compliance with a special diet.
Children perceive oral speech well and understand the content of almost all conversations, but rarely respond. They often refuse to take part in the conversation and use several dozen words in their speech.
Symptoms
The first signs of Angelman syndrome appear towards the end of the child's first year of life. Symptoms become more obvious after reaching the age of two. The syndrome is characterized by polymorphic clinical manifestations. Patients can be recognized immediately. Such children have a characteristic appearance.
- Features of the appearance of a sick child: discrepancy between a small head and a normal body, large mouth, constant smile, wide interdental spaces, narrow lips, protruding wide tongue, malocclusion, flat back of the head, smooth palms, pointed chin, hypopigmented skin, strobism, scoliosis, walking on straight legs.
- Violation of psycho-emotional development is manifested by mental retardation, excessive fussiness, friendliness, causeless laughter, and euphoria.
- Neurological symptoms: limb tremors, ataxia, incoordination, loss of balance, muscle hypotonia, sleep disorder, hysteria, speech impairment, hyperactivity and hyperexcitability, learning difficulties.
- Movement disorders: poor control over the movement of the tongue and its unreasonable protrusion, difficulties with swallowing and sucking, arms raised or bent while walking, frequent drooling, irrepressible thirst, excessively active chewing movements, impaired fine motor skills, epilepsy.
Children with Angelman syndrome are prone to nonverbal communication: they perceive oral speech, but cannot express their thoughts. They practically do not participate in the conversation, since their vocabulary consists of 10-20 words necessary in everyday life. Patients have stereotypies - repeated flapping of arms, twisting of hands and frequent clapping of hands, as well as other actions that are not typical for healthy people. A shaky gait is accompanied by sharp twitching of the arms, which makes the person look like a puppet. Patients feel more comfortable in water and do not tolerate stuffy rooms and high temperatures.
With age, the clinical picture of the disease changes significantly. Convulsions and seizures occur less and less often or disappear completely. Patients become calmer and their sleep improves. Men and women with this syndrome have a youthful appearance that hides their true age. They experience puberty on time, and may even have children.
Symptoms of the pathology in adults include impaired fine motor skills and urinary incontinence. Patients cannot cope with buttons and zippers on clothes, but at the same time they can easily use a fork and spoon. They are friendly, loving and affectionate. Obesity is a common disorder among adult patients, especially women. People with Angelman syndrome are prone to autonomic and somatic disorders. They do not tolerate hot weather well and often suffer from constipation and esophageal reflux.
Since the disease is congenital, all of these anomalies can appear immediately after childbirth. Modern diagnostic methods make it possible to identify the syndrome in utero and prevent the birth of a sick child.
Video: examples of children with Angelman syndrome
Diagnosis and timely detection
Doll syndrome can be diagnosed before the baby is born by conducting a genetic study of the fifteenth chromosome.
Diagnosis can be invasive or non-invasive. An invasive research method has existed in medicine for a long time, but is quite risky, since it requires penetration into the uterus and collection of amniotic fluid.
The non-invasive method involves testing the mother's blood and testing the baby's DNA. Based on this study, conclusions are drawn about the presence or absence of certain deviations.
Diagnosis is also carried out on newborns who exhibit disturbances in muscle tone and delays in the development of speech and motor skills.
It is necessary to monitor the baby’s facial expression, behavior, expression of emotions, and movements. If a baby has difficulty bending the limbs, tremors, chaotic and sudden movements of the limbs are observed, then it is worth seeking advice and help from specialists.
Often, the diagnosis of Angelman syndrome is made between the ages of 3 and 7 years, when a clear manifestation of the signs of this disease can be noticed. Depending on the extent of damage to chromosome 15, manifestations can vary in severity: some patients even have difficulty speaking, while others can lead an independent life.
Necessary diagnostics
Confirming the presence of a problem begins with taking a history and examining the child. As a rule, suspicion of Angelman syndrome arises due to the specific behavior and appearance of the patient. However, to make an accurate diagnosis, genetic tests will be required. The examination of the child also includes standard blood tests, which make it possible to exclude other possible disorders that are accompanied by the development of a similar clinical picture. Computed and magnetic resonance imaging is also widely used to obtain layer-by-layer photos of bone structures and the brain. Such studies are necessary in the presence of neurological symptoms.
Photo and video materials on the topic
Video, as well as a gallery with photos of children and adults diagnosed with Angelman Syndrome:
Child with doll syndrome Adult with Angelman syndrome
Causes
The causes of Angelman syndrome are defects in various genes of the fifteenth chromosome. Also, this disease can appear due to a mutation in the mother's chromosome (mutations in the father's gene provoke the development of Prader-Wili disease).
Basically, parsley syndrome is associated with the loss of a certain part of the chromosome or with mutational changes in the UBE3A gene.
The spontaneous development of defects in chromosome 15, also causing parsley syndrome, is associated with the complete absence of four million base pairs of DNA.
Improving the condition and life of patients
Angelman syndrome is a genetic disease; today there are no methods for restoring chromosomal abnormalities, and treatment is impossible. However, there are a large number of ways to reduce symptoms, which makes the condition of such people easier.
In each specific case, the rehabilitation program is developed individually, according to the symptoms and condition of the individual patient. Experts identify four main areas of therapy:
- Taking antiepileptic drugs and anticonvulsants . These medications help control and reduce the frequency of attacks caused by the disease.
- Therapeutic exercise – helps develop fine motor skills and solve other problems of the musculoskeletal system. Children with chromosome abnormalities develop more slowly than their healthy peers, and this requires patience on the part of the family.
- Sign language . Patients with marionette syndrome speak little, but use sign language quite successfully. Education should start from a very early age.
- Behavioral therapy . This program allows you to provide correct and effective education to children who have disabilities and will help cope with hyperactivity and attention deficit.
Many doctors note the similarities between children suffering from autism and Angelman syndrome. American scientists have achieved success in treatment, namely the use of intravenous injections with the hormone Secretin. The positive effect is expressed in reducing signs of unwanted behavior, as well as improving communication skills.
Treatment of Angelman's disease
Today, therapy for Angelman syndrome is aimed at eliminating/mitigating symptoms and is supportive in nature. There are no specific drugs or genetic correction methods that can cure the disease. In most cases, patients with this syndrome are in relatively good general physical health, and children can receive standard pediatric care, including immunizations.
In cases of seizures, the use of anticonvulsants is recommended. As a rule, such manifestations of the disease can be adequately controlled with a single drug. However, sometimes relief of such symptoms requires a combination of several medications.
It is important to note that there is no single anticonvulsant drug that is effective for most cases of Angelman syndrome.
Therefore, it is necessary to consult with your doctor, who will prescribe the most effective remedy/set of remedies, based on the individual characteristics of the patient.
Features of education and adaptation
As a child with this pathology grows up, the symptoms change, become less intense, or, on the contrary, more intense, old ones may disappear and new ones may appear.
The prospects for the patient's condition largely depend on the conditions that surround the patient. A friendly environment, an atmosphere of love and affection gives the patient a chance to become at least partially an independent person. With proper care, the symptoms of the disease become easier over time. Life expectancy for people with Angelman syndrome is average. It is necessary to remember about regular examinations, proper nutrition, and compliance with treatment conditions.
Educating a child at school requires the use of inclusive programs. However, in Russia and the CIS countries today these programs are not widely practiced. Such children need a special approach, including learning to concentrate. An important requirement is also the ability of the educational institution to provide medical assistance during a seizure.
In our society at the moment, people with chromosomal disorders are treated with caution and fear. In recent years, various events have been actively carried out aimed at disseminating knowledge about such diseases, as well as at developing tolerance in society.
Forecast
The prognosis of the pathology is determined by the degree of damage to chromosome 15. Patients with minor changes can take care of themselves and behave normally in society. Children with a pronounced genetic defect do not speak and practically do not walk. The disease is most severe in children whose chromosomes lack gene sections. They usually cannot walk or talk on their own. All other forms of pathology can be corrected. In any case, such patients will not be able to become full-fledged members of society. They understand speech, but do not engage in dialogue. They are taught sign language from childhood. Verbal communication is not available to them.
Patients with Angelman syndrome require special attention and care. Relatives and friends should create an atmosphere of understanding, love and friendliness for the patient. Sick children need help adapting to life in society. They are shown a special diet and sleep regimen, careful care, and training in basic self-care skills. There are specialized boarding schools for such children, which provide a special course of adaptation to society and training. Patients are taught sign language and undergo special programs aimed at developing fine and gross motor skills.
With age, hyperactivity and sleep disturbances go away on their own. If treatment is started early and carried out competently, the prognosis of the pathology improves. The disease does not affect life expectancy.
With proper treatment and careful attention, life can be made much easier for people with Angelman syndrome, but they will never be able to live independently. This is the category of people who will constantly need guardianship and care.
External signs
Children susceptible to the disease exhibit special external facial features that resemble the face of an elf. This is where another name given to this disease comes from - Elf syndrome, because it looks like an elven face from literary sources.
External signs of Williams Syndrome
It is important to consider the following external signs:
- broad forehead;
- divergence of eyebrows along the midline;
- eye color is bright blue;
- cheeks full and drooping;
- thin upper lip and full lower lip
- wide mouth, smile from ear to ear;
- flat bridge of the nose;
- the nose is peculiar with a flat, blunt end;
- The chin is small and somewhat pointed.
These signs are changeable, but their combination indicates the manifestation of mutations. Over time, appearance may change: an increase in the brow ridges, a decrease in facial pastiness, a flat bridge of the nose disappears, but the distance from the upper lip to the tip of the nose remains very noticeable.
Prevention
It is impossible to prevent the birth of a child with Angelman syndrome, since gene mutation occurs spontaneously. Even the most qualified geneticists cannot explain such a process.
Preventive measures are aimed at thorough preparation for pregnancy and childbirth. All pregnant women must undergo a fetal ultrasound at the appointed time and donate blood for serum markers of chromosomal pathologies. In case of a positive result of a medical genetic study, parents must decide whether they can raise a sick child.
Medical genetic counseling is required for couples with a family history. If this disease has not been recorded in loved ones, it is enough to follow the basic principles of maintaining a healthy lifestyle during pregnancy.
Angelman syndrome is a severe and incurable disease that deprives a person of a normal life. Sick children need special care and attention. Treatment is aimed at reducing the intensity of the symptoms of the pathology. Modern scientists are trying to find effective ways to combat chromosomal abnormalities that affect newborn children.
Development of children with Williams syndrome
Teaching children with Williams syndrome requires an individual approach. The pedagogical process is associated with the formation of correct speech, learning to write and read. Patients with the syndrome have a difficult time learning basic skills. Children have a reduced level of attention in the process of mastering educational material.
Mathematical subjects are difficult for them, but they quickly master reading. Many have a good ear for music. Mutual understanding and friendliness in the family play an important role in the treatment process. Psychotherapy and sessions with a psychologist become an integral part of the treatment, which helps children better undergo the socialization process.
Notes
- Monarch Disease Ontology release 2018-06-29sonu - 2018-06-29 - 2018. https://wikidata.org/Track:Q55345445″>
- X-linked mental retardation. (Russian) (inaccessible link). Center for Molecular Genetics at the Medical Genetic Research Center of the Russian Academy of Medical Sciences. Retrieved May 8, 2019. Archived June 4, 2016.
- Yamasaki K., Joh K., Ohta T., Masuzaki H., Ishimaru T., Mukai T., Niikawa N., Ogawa M., Wagstaff J., Kishino T.
Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. (English) // Human molecular genetics. - 2003. - Vol. 12, no. 8. - P. 837-847. — DOI:10.1093/hmg/ddg106. - PMID 12668607. - Harry Angelman (English) // Wikipedia. — 2018-10-28.
- Harry Angelman.
'Puppet' Children A Report on Three Cases (English) // Developmental Medicine & Child Neurology. — 2008-11-12. - Vol. 7, iss. 6. - P. 681–688. — ISSN 1469-8749 0012-1622, 1469-8749. — DOI:10.1111/j.1469-8749.1965.tb07844.x. - Story. Dr Harry Angelman, a pediatrician who worked in Warrington (formerly Lancashire), first reported three children with the condition in 1965 and specifically
. medlec.org. Retrieved November 22, 2019. - Petersen MB, Brøndum-Nielsen K., Hansen LK, Wulff K.
Clinical, cytogenetic, and molecular diagnosis of Angelman syndrome: estimated prevalence rate in a Danish county. (English) // American journal of medical genetics. - 1995. - Vol. 60, no. 3. - P. 261-262. — DOI:10.1002/ajmg.1320600317. - PMID 7573182. - Steffenburg S., Gillberg CL, Steffenburg U., Kyllerman M.
Autism in Angelman syndrome: a population-based study. (English) // Pediatric neurology. - 1996. - Vol. 14, no. 2. - P. 131-136. — DOI:10.1016/0887-8994(96)00011-2. - PMID 8703225. - Angelman Syndrome (English) (undefined)
. NORD (National Organization for Rare Disorders). Retrieved September 19, 2019.
Drug treatment
It is important to remember that the most important thing for children with this disease is an atmosphere of love and affection in the home; do not forget about regular examinations and compliance with all conditions of treatment therapy. Over time, new treatments for Angelman syndrome are being developed. These special programs are compiled individually for each individual patient with different symptoms, but there are 4 main methods:
- Anticonvulsant medications for the treatment of epilepsy, which will reduce the risk of frequent seizures and ease the consequences.
- Therapeutic exercise for the development of fine motor skills and other types of physical activity.
- It is better to start learning sign language from an early age; learning this method of communication will help you quickly adapt and socialize, as far as possible in this case.
- Behavioral therapy is necessary to overcome hyperactivity and improve concentration.
Use medications strictly as prescribed by your doctor, since self-medication can be fraught with serious side effects and complications. The main complications are considered to be deterioration of the condition and the spread of atony to other areas. Consultation with a doctor is the main precaution that will ensure the safety of treatment, make it as effective as possible, and avoid side effects.
If a decrease in muscle tone is accompanied by severe pain, painkillers should be taken. Paracetamol, which is taken one tablet three times a day, has worked quite well.
For children under 7 years of age, when pain occurs, it is recommended to take Panadol, 1 teaspoon (4-5 measuring spoons) per day.
Also, for the treatment of pain in combination with spasms and convulsions, it is recommended to use cabrazepam at a dose of 3-5 mg/kg body weight 2-3 times a day.
For severe pain, ketonal is recommended - 50 mg 1-2 times a day, maximum daily dose 100-150 mg.
Mydocalm
It is a drug that is prescribed primarily to normalize muscle tone. A characteristic feature is that it optimizes tone. In case of hypertonicity, it helps to reduce the tone of tense areas; in case of atony, on the contrary, it tones the muscle. Also prescribed for radiculitis, arthritis, spondyloarthritis, lumbodynia, osteochondrosis.
Normalizes metabolic processes in muscle tissue and joints. It relieves pain well, eliminates spasms, and promotes recovery processes. Prescribed for recovery after various operations, especially when restoring the musculoskeletal system. It is also effective in the treatment of ulcers, erosions, erysipelas, and is used for wound healing.
Available in the form of tablets and solution for injection. Used intramuscularly in the form of injections of 100 mg, 2 injections per day. If the drug is prescribed in tablets, 150-450 mg per day is recommended (for adults).
Corn oil or any other fatty base is used as the basis for preparing a decoction for medicinal baths. Heats up to a warm state. It is better to use a water bath. 2 ml of concentrated extracts of the following plant components are poured into the resulting oil: common lilac, buckthorn bark, lemon balm leaves, valerian root, cinquefoil herb. Used for massage and rubbing.
Take any body cream. It is better to take natural cream, without adding impurities, flavors, or dyes. Even baby cream will do. Add a tablespoon of water or alcohol infusions of cinnamon, cloves, chamomile flowers, and calendula flowers to it. You can add 1 ml of water infusion of hot red pepper.
For general strengthening of the body, use balm. It is prepared on the basis of cognac. To prepare, take 2 tablespoons of valerian herb, Manchurian aralia rhizomes, calamus rhizomes, caraway fruits, chamomile and a tablespoon of honey. Pour in cognac and leave for at least a day. Drink 10 grams per day.
Let's look at some recipes that are widely used to treat the lumbar spine.
To prepare, take a tablespoon of concentrated herbal extract of hernia, black poplar, 50 ml of chilibuha bark tincture, a teaspoon of eucalyptus oil, 10 ml of fresh ectericide. All this is infused for at least 3-4 days, drinking 50 ml per day.
Take equal parts of mistletoe, drop cap leaves, wormwood branches, triphol leaves, black elderberry flowers (at the rate of one dessert spoon per 500 ml of alcohol or cognac). Insist for at least 5 days, drink in small quantities twice a day, 28 days.
Vodka or pure alcohol is used as a base. Then add about a tablespoon of the following components: formic alcohol, alcohol tincture of chestnut fruits, 5 drops of henbane alcohol tincture, a teaspoon of camphor oil. Add a tablespoon of rose hips and a teaspoon of citvar seed. Stir until a homogeneous consistency is formed, then set aside and allow to harden.
To ordinary alcohol (500 ml) add a tablespoon of common wormwood, Damask rose flowers, and tansy. Then add 2-3 drops of essential oil of thuja and juniper. Drink a tablespoon twice a day.
Strengthening and stimulating balms prepared on the basis of herbal remedies, as well as components of animal origin, have proven themselves well. Let's look at some recipes used in the treatment of cervical muscular-tonic syndrome.
To prepare, take a tablespoon of honey, clover roots, dandelion roots and grass, nettle and buckwheat leaves. All this is infused for at least 3-4 days, drinking 50 ml per day. The course of treatment is at least 28 days (full biochemical cycle).
Take equal parts of fireweed leaf, yarrow flowers, 10 ml of horseradish juice, 2 tablespoons of vinegar, 2 drops of iodine. Insist for at least 5 days, drink in small quantities twice a day, 28 days.
Vodka or pure alcohol is used as a base. Then add about a tablespoon of the following components: calamus, dandelion, wormwood, blue cornflower. Add a tablespoon of fresh black radish juice, a tablespoon of horseradish puree, 10-20 mustard seeds. Stir until a homogeneous consistency is formed, then set aside and allow to harden.
To ordinary alcohol (500 ml) add a tablespoon of the ground part of the thistle plant, the juice of half an orange, a teaspoon of celandine juice, 50 ml of 96% alcohol tincture of birch buds and 5-6 dried chestnut flowers. Then add 2-3 drops of essential oil of coriander and lemongrass. Drink a tablespoon twice a day.
To eliminate thoracolumbalgia with muscular-tonic syndrome, a complex effect is needed. To do this, you need to consult a doctor. But in order to quickly relieve pain, you can try rubbing the painful area and applying ointment.
To prepare the ointment, take lard as a base, melt it in a water bath, or over low heat until dissolved, with constant stirring. Add 2 tablespoons of bell flowers, birch branches, Leuzea root, plantain root, and 25-30 drops of Eleutherococcus infusion to the resulting mass.
As a basis for preparing massage oil, take approximately 100 ml of a mixture of burdock, castor and olive oils. A mixture of the following plant components is prepared in advance in a fireproof container: caraway seeds, Siberian rowan fruits, honey, rose hips, strawberry leaves (at the rate of approximately 2 tablespoons of each component per 100 ml of oil).
As a base, take a mixture of massage base oils: peach seed oil, wheat germ oil in a ratio of 1:2, add 2-3 drops of castor and sea buckthorn oils. Stir. Add 2 drops of the following essential oils to the resulting mixture: nettle, birch, orange. Also add 50 ml of aloe juice, 50 ml of Cahors and 5 grams of gelatin. Mix thoroughly and use during massage.
Lanolin cream is used as a base for preparing massage oil. 2 ml of concentrated extracts of the following plant components are poured into it: parsley root, dried seaweed, hazelnut kernels freed from scales. Used for massage and rubbing.
Take any body cream. It is better to take natural cream, without adding impurities, flavors, or dyes. Even baby cream will do. Add 150 ml of aloe juice, 150 grams of honey, 10 grams of dry gelatin, one raw egg to it. Mix all this until smooth, you can lubricate painful areas with this cream before going to bed.
[1], [2], [3], [4], [5], [6], [7]
Flax seed allows you to restore tone, normalize metabolic processes, increase elasticity, conductivity and resistance of muscle tissue and nerve fibers. It is useful to drink a decoction of the seed instead of water. First, the seed should be fried in a frying pan until it turns brown, then grind it in a coffee grinder and brew it like coffee. You can add honey and lemon to taste.
For atony, it is recommended to take a decoction of birch buds internally. Quickly relieves spasms, eliminates pain and restores tone.
Rue officinalis root in the form of a decoction (a teaspoon in a glass of water) is taken for 28 days to restore muscle tone.
The main methods of treatment are treatment with ultrasound, microcurrents, and waves of various lengths. Electrophoresis is used, with which drugs are injected directly into the damaged tissue. Acupuncture, also known as acupuncture, is a fairly effective method.
Puppet Theatre
Parsley Syndrome (2015) drama Director: Elena Khazanova Cast: Evgeny Mironov, Chulpan Khamatova, Merab Ninidze, Vladimir Seleznev, Alexander Kuznetsov Premiere: November 5, 2019 Watch in the service
Since childhood, Petya has been in love with the neighbor's girl Lisa, he spends his evenings with her as a child, runs away with her from his parents' house to St. Petersburg, and against the will of his father, Lisa becomes his wife and main life partner. But Peter also has another passion - from the same young age he has been obsessed with puppet theater and puppets, the stage largely replaces real life for him, and the artist cannot separate art and family. The death of the newborn first child of Peter and Lisa falls heavily on the lovers, the woman even has to go to a clinic for the mentally ill. But Peter is more likely to need the help of a psychiatrist - in Lisa’s absence, he makes an exact copy of her, a doll that will become his real soul mate.
Still from the film “Parsley Syndrome”
Elena Khazanova dreamed of working with Chulpan Khamatova back in 2006 on the film “Play on Words: The Oligarch’s Translator,” but then the main role went to Yulia Batinova, because Khamatova did not speak French
Against the background of films of the light genre, real serious dramas have somehow completely disappeared in Russian cinema; the release of almost every film that goes beyond romantic comedies or youth adventures becomes a small event, at least for the industry and critics. Elena Khazanova’s “Parsley Syndrome” was eagerly awaited by the director’s colleagues and journalists, and the premiere at the Kinotavr festival did not leave specialists indifferent, but now the film faces a new test - the film is being released in wide release, which means it will receive appreciation from viewers and fans of Dina’s work Rubina and Evgeniy Mironov. And we can predict some questions for future viewers about the film today.
Still from the film “Parsley Syndrome”
At the post-production stage, production companies from Switzerland and Germany joined in financing the film, which means that “Parsley Syndrome” will receive distribution at least in these European countries
We believe that the film will cause the greatest resonance among fans of Rubina’s work - her books are rarely filmed, and even more rarely do these films manage to break out beyond a small niche audience. “Parsley Syndrome,” made for the widest possible public, may nevertheless also be deprived of attention; it is aimed at a more mature, intelligent and thinking audience than the one that fills cinema halls during screenings of Hollywood blockbusters. It will not be easy to attract viewers, although the participation of Evgeny Mironov and Chulpan Khamatova may add advantages to the film's distribution karma.
Still from the film “Parsley Syndrome”
Another obstacle on the way of “Syndrome” to the viewer may be the assessments of those very fans of the writer, because the script of the film significantly simplifies, truncates and unifies a huge literary canvas. The plot has lost almost half of the characters, the emphasis has been placed on only a couple of storylines, time periods have been shuffled - all this invariably causes grumbling among the followers of the teaching “The book is better!” Of course, comparing a film and a book is a thankless task, but it is rarely possible to do without it, and we dare to assume that there will be plenty of literary copies around “Petrushka” that will be broken.
Still from the film “Parsley Syndrome”
Fans of Yevgeny Mironov will face no less debate. Of course, in a complex psychological drama, this talented actor feels like a fish in water, but Evgeniy has long been haunted by one creative ailment - he is overly theatrical. Even in this film, where he plays a man obsessed with art, the stage, and acting, Mironov often bends, wrings his hands, and turns the face of the film into a grimace. Often these hyperboles are unnecessary; they complicate the action and distract attention from the internal nerve to external emotions.
Still from the film “Parsley Syndrome”
However, as you understand, these are only small claims, largely far-fetched and sucked from the fingers - on the whole, Khazanova’s picture turned out to be surprisingly holistic, deep and unusually touching. Even the gloom and grayness, illuminated only by the lush red head of hair of the heroine Khamatova, turn out to be “suitable” for “Syndrome”. No matter what mystical notes are attributed to it, no matter how many “come to life” dolls appear in the frame, the film is, first of all, a heavy psychological drama about the struggle of opposites. Here real life collides with fictional theatricality, ancient curses collide with modern medicine, passion and reluctance to let go confront freedom, love borders and sparkles from contact with hatred.
Following the relationship between a couple of literally madly in love characters, Mironov and Khamatova, is incredibly exciting, the open ending leaves the opportunity to interpret the present and, especially, the future of the characters in a variety of ways, and most importantly, with all its focus on the world of creative people, the plot and the vicissitudes of the life path of Peter and Lisa can easily be apply to the surrounding reality. The madness of passion is closer than we think, and resisting it is more difficult than we think, and Parsley-Pygmalion syndrome is not an amazing hereditary disease that occurs once in a million, but almost a general epidemic that locks people in the worlds of puppets and weak-willed dolls.
From November 5th in cinemas.
What do elf children mean?
“Elf children” or Williams syndrome is a genetic disorder characterized by the absence of several genes in the human genome. The total number of genes in such patients can range from 25–29. Depending on the number of missing units of genetic material, the symptoms and severity of pathological changes change. The disease was first described in the 60s of the 20th century. At this time, the hereditary nature of the disease has been fully established. According to geneticists, the pathology is the result of a spontaneous mutation in the body.
Williams syndrome - type of inheritance
In the course of lengthy laboratory studies, geneticists were able to establish that elf children are born as a result of a deletion of the 7th chromosome near its long arm. The length of the missing section can vary between 1.6–1.8 million nucleotide pairs. A disturbance in the composition of the gene apparatus is provoked by a spontaneous mutation.
However, there is evidence indicating an autosomal dominant type of inheritance of the pathology. Compared to other genetic hereditary pathologies, Williams syndrome is a common disease: the incidence can reach 1 case per 20,000 newborns. At the same time, there is no pattern of gender distribution of the disease: girls and boys are affected by the disease with the same frequency.
Causes of Williams syndrome
As the results of genetic studies have shown, elf children (Williams disease) are born as a result of a mutation occurring at the stage of meiosis. Chromosomes are damaged during the formation of sex parent cells. If one of them has an anomaly, there is a high probability of abnormalities developing in the embryo. The absence of the ELN gene on the chromosome causes insufficient elastin formation.
This substance is part of the walls of large blood vessels and heart valves. As a result of such changes, patients with Williams syndrome develop pathologies of the cardiovascular system, heart defects, and arterial hypertension. The absence of the LIMK1, GTF2IRD2 genes causes pathological changes in neurons, which result in neurological abnormalities.
Children with Angelman syndrome: adaptation and education
Children with doll syndrome need more attention than healthy babies. A sick child is usually hyperactive, more drawn to people, loves to play and communicate.
Characteristic signs in behavior and communication:
- Excessive friendliness;
- Good nature;
- Attachment to people.
Mental retardation may not be very pronounced - children distinguish emotions well enough and understand others. But at the same time, such children are very gullible and do not understand well when they are being deceived.
Due to an atrophied sense of satiety, they may suffer from obesity.
The disease does not affect life expectancy. Parents need to help such children adapt, follow a diet and sleep schedule that may cause problems. Most of these children master all self-care skills as they grow up.
Muscular-tonic syndrome in a child
A decrease in muscle tone in children can be caused by both congenital factors and external factors. Thus, congenital factors are most often incurable and are genetically determined. It is noted that such conditions often progress and end in paralysis and disability.
Most often, a child’s muscle pain is associated with the so-called “disease” of growth, that is, the symptom is caused by a completely normal, natural process of growing up. Some children do not feel discomfort associated with growth at all, while others react quite painfully. The etiology of myalgia in children is not fully understood, but the generally accepted version is a discrepancy between the rates of growing bone and muscle-ligamentous systems. The skeleton grows faster, tendons and muscle tissue do not have time to adapt to the speed and intensity of growth.
Of course, this explanation is extremely simplified; in fact, in a child’s body everything is more complicated. It is believed that muscle pain in a child is associated with hidden congenital or acquired chronic pathologies. The most common muscle pain symptom is in children aged 3.5-10 years; adolescents also suffer from myalgia, but it has a more precise etiological cause.
Muscle pain can be a symptom of an underlying disease, or less often it is an independent condition.
List of factors and conditions that cause reversible muscle pain in a child:
- Cramps, which may be a consequence of “growing pains” or caused by a sports injury, bruise, or torn ligaments.
- The inflammatory process in muscle tissue is myositis, provoked by viral pathologies (influenza, acute respiratory infections), bacterial infection, including parasitic ones. The pain is localized in the large muscles of the body - in the back, shoulders, neck, arm muscles.
- Dehydration during active physical activity, which is typical for children who are fond of sports games in the hot season. Loss of fluid through sweat leads to a deficiency of magnesium and potassium, and hyperventilation during fast running can lead to cramps in the calf muscles.
In addition, there are a number of serious pathologies that are characterized by muscle pain in children:
- Duchenne myopathy. This is a pathology diagnosed in boys in early childhood. The disease has a genetic cause - an abnormality of the X chromosome. The consequence is a gene mutation and dystrophin protein deficiency. Pseudohypertrophy develops slowly and gradually affects all skeletal muscles, less often the myocardium. The clinical picture is determined at the age of 3-4 years, when the baby has difficulty climbing stairs and cannot run. The prognosis of the disease is unfavorable.
- Becker pseudohypertrophy is a disease similar to Duchenne myopathy, but weaker in clinical manifestations and more favorable in course and prognosis.
- Bornholm's disease or epidemic myalgia. The disease is viral in nature (Coxsackie virus), develops rapidly, accompanied by severe muscle pain in the chest, less often in the abdomen, back, arms or legs. The disease is diagnosed by specific symptoms - fever, myalgia, vomiting. The pain is paroxysmal, subsides at rest and intensifies with movement. Epidemic myalgia quite often coexists with enteroviral infections, herpes, and serous meningitis.
Fibromyalgia and polymyositis (dermatomyositis) do not occur in children; isolated cases are so rare that they are considered a diagnostic phenomenon or an error.
Thus, unlike adults, 85-90% of muscle pain in children is caused by physiological or situational factors. Such pain can be defined as a curable, reversible symptom. However, if pain prevents the child from moving normally, is accompanied by hyperthermia, visible bodily defects (curvature, protrusion, depression), parents should urgently consult a doctor to examine the child and begin adequate treatment.
Treatment of Williams syndrome
Williams syndrome, "elf face" is a genetic pathology, so it cannot be cured. Doctors direct therapeutic measures to improve the patient’s well-being and improve his living conditions. Surgery is performed to correct congenital heart defects in infancy. To combat severe arterial hypertension, antihypertensive drugs are used. Psychocorrectional work is actively carried out, which helps children master speech, writing and reading, and reduces the possibility of mental retardation.
Parsley syndrome: illness and life prognosis
Life prognosis and severity of the disease depend on the severity of symptoms. In each specific case the situation may be different.
So some children with a milder form of the disease may:
- Be able to control yourself;
- Process information better;
- Have a large vocabulary smell.
In another case, the child may have more problems with intelligence, but at the same time be well physically developed. The severity of the disease is influenced by whether there are epileptic seizures or not.
Early initiation of restorative therapy and the use of physical procedures significantly reduces the manifestation of the disease and improves life prognosis.
There are also genetic studies that indicate that a certain type of mutation in genes has different effects on the diagnosis and course of the disease. Thus, changes in the UBE3A gene lead to the least changes in the body, while large rearrangements in chromosome 15 can greatly affect the condition of a sick child.